铁死亡(Ferroptosis)是一种铁依赖的磷脂过氧化驱动的新型细胞死亡形式。越来越多的证据表明,铁死亡在包括癌症、缺血性器官损伤和退行性疾病等多种病理条件中发挥着重要作用。铁死亡诱导作为一种潜在的癌症治疗方法,无论是作为单一药物疗法还是与其他靶向药物(包括PI3K/mTOR通路抑制剂和免疫检查点阻断剂)联合应用,都显示出非常有前景的应用价值。因此,探讨铁死亡的分子机制对于基础生物学和疾病治疗都具有重要意义。
磷脂过氧化会在细胞正常代谢和应激反应中自发产生,因此生理条件下,细胞需要监测机制(surveillance mechanisms)来抵抗由磷脂过氧化引发的非预期性铁死亡。目前铁死亡监测主要有两种已知的调控机制:1)通过谷胱甘肽过氧化物酶4(GPX4)介导磷脂过氧化物(PL-OOH)还原为相应磷脂醇(PL-OH);2)通过FSP1、DHODH、NOS2和GCH1等酶产生具有俘获自由基能力的抗氧化细胞代谢产物(radica-trapping antioxidant,RTA,比如CoQ10, NO, BH4),从而终止磷脂过氧化以阻断铁死亡。研究发现,这些监控机制在多种肿瘤细胞中呈现活跃状态,癌细胞可能利用这些机制来逃避铁死亡。如何通过抑制这些监测机制来实现诱导铁死亡,是目前肿瘤治疗领域的研究热点。发现独立于GPX4和RTA意外的铁死亡监测机制,对于开发针对诱导肿瘤铁死亡的联合治疗策略具有重要的指导意义。
2023年6月1日,来自纪念斯隆-凯特琳癌症中心的姜学军团队在Cell发表题为Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones的文章,揭示了一种受性激素调控但不依赖于GPX4的铁死亡新监测机制。

为了鉴定新的铁死亡抑制基因,作者使用GPX4抑制剂RSL3和Cystine starvation作为铁死亡诱导剂,对人成纤维肉瘤细胞HT1080中进行了全基因组CRISPR的激活筛选,结果显示多个基因的sgRNA在铁死亡诱导条件下的存活细胞中显著性富集。在富集最高的7个基因中,有5个已知的铁死亡抑制基因:SLC7A11,NFE2L2/NRF2,FSP1,GCH1和NOS2;另外两个是未被报道有铁死亡调控功能的脂质修饰酶MBOAT2和PLA2G2F(图1)。

接下来,作者在体外实验中验证了MBOAT2和PLA2G2F的过表达均能显著性抑制RSL3或者GSH耗竭引发的铁死亡。进一步,作者发现MBOAT2而非PLA2G2F能显著抑制HT1080-GPX4KO细胞的自发性铁死亡。不仅如此,MBOAT2还有效抑制了GPX4/FSP1DKO细胞的自发性铁死亡,并保持了长期的细胞存活。这一结果表明MBOAT2是一种不依赖于GPX4和FSP1的强效铁死亡抑制基因。
作为膜结合O-酰基转移酶(membrane bound O-acyltransferse, MBOAT)家族的成员,MBOAT2具有溶血磷脂酰基转移酶(lyso-PL acyltransferase,LPLAT)活性。已有对于MBOAT2的功能研究很少,文献提示MBOAT2偏好将单不饱和脂肪酸(MUFA)转移至Lyso-PL并生成PL-MUFA。作者通过实验证实MBOAT2可以利用从头合成途径生成的MUFA或者摄取的外源性MUFA来介导抑制铁死亡的功能。敲减MBOAT2可以显著降低MUFA对于铁死亡的抑制功能。通过脂质组学分析,作者发现MBOAT2过表达选择性增加含有单不饱和脂肪酸的磷脂酰乙醇胺(PE-MUFA)的生成,并同时选择性降低含有多不饱和脂肪酸的磷脂酰乙醇胺(PE-PUFA)的生成。这些结果提示,MBOAT2具有MUFA和Lyso-PE(溶血磷酯酰乙醇胺)双重偏好性。于是,作者提出了一个竞争性的PE重塑模型,在该模型中MBOAT2作为偏好MUFA的溶血磷酯酰乙醇胺酰基转移酶(Lyso-PE acyltransferase, LPEAT)与偏好PUFA的LPEAT(比如LPCAT3)竞争Lyso-PE,在MBOAT2具有主导优势的特定细胞中,PE重塑将使细胞处于高PE-MUFA水平但低PE- PUFA水平的状态,从而导致铁死亡的抗性,反之亦然。
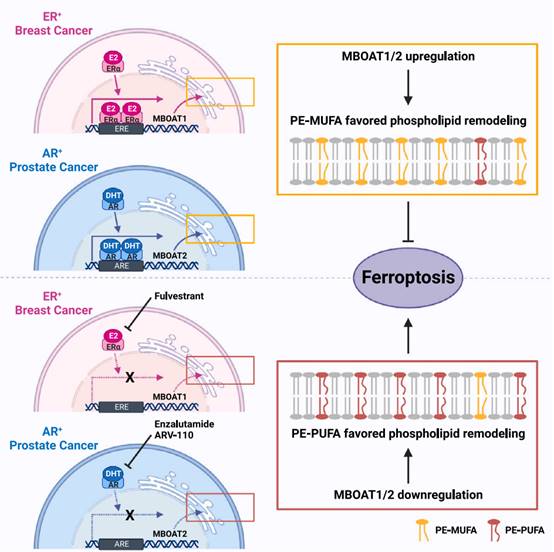
进一步,作者发现MBOAT2在前列腺癌(PCa)中呈现特异性上调(TCGA PanCancer Atlas),且人类PCa样本中MBOAT2与AR的mRNA表达呈正相关,通过一系列实验作者证实了MBOAT2是前列腺癌细胞中AR的真正靶点。接着,作者进一步检测了MBOAT2对AR阳性PCa细胞的铁死亡敏感性的影响,发现AR阳性比AR阴性的PCa细胞系更耐受铁死亡,这一调控是通过上调MBOAT2表达来重塑细胞磷脂组分来实现的。进一步,作者发现恩扎鲁胺(ENZ,第二代FDA批准的针对AR的治疗药物)与ARV-110(一种临床阶段的AR降解剂)均能显著性抑制内源MBOAT2表达,并显著增加AR+PCa细胞对RSL3诱导的铁死亡敏感性。尤为重要的是,在LnAR-iCas9-gGPX4(LnAR-igGPX4)的移植瘤动物模型中,多西环素(Dox)饲料(Dox诱导Cas9表达后,可以敲除细胞中的GPX4基因)与ENZ的联合使用完全抑制了肿瘤生长,这表明ENZ与铁死亡诱导的联合治疗是治疗AR+PCa一种潜在方案。
有趣的是,作者发现与MBOAT2类似,这一家族的另一成员MBOAT1亦可通过调控PE- MUFA偏好的PE重塑机制抑制铁死亡。但又与MBOAT2不同的是,MBOAT1更多是在女性癌症中呈现高度表达,如卵巢癌、乳腺癌和子宫内膜癌(TCGA PanCancer Atlas),提示MBOAT1极有可能受雌激素受体(ER)信号的调控。接着,作者证实了MBOAT1在ER阳性乳腺癌中被ER特异性上调,且对ER阳性乳腺癌的铁死亡抵抗有重要贡献。此外,作者惊喜地发现:Fulvestrant(Ful,一种ER抑制剂)耐药的MCF7细胞系(MCF7-FulR+)保留了Ful对ER的降解和对MBOAT1下调的作用,这一现象促使作者思考:ER抑制剂与铁死亡诱导联合治疗是否是一种潜在的治疗ER+乳腺癌耐药的疗法?接下来的MCF7-FulR+异种移植动物模型结果显示,Ful和IKE(一种可用于动物实验的铁死亡诱导剂)的联合应用显著抑制了肿瘤生长,提示ER靶向治疗与铁死亡诱导的联合治疗可能让ER抑制剂耐受的ER阳性乳腺癌患者受益。
综上所述,这一系统性的研究揭示了一种调控铁死亡监测的新分子机制,即性激素信号可以通过MBOAT1/2介导的磷脂重塑抑制癌细胞铁死亡(图2),这是一种不依赖于GPX4和FSP1的预防铁死亡的新监测机制,这一发现对整个铁死亡领域有着重要意义,并且在癌症研究方面具有重要意义,可能为开发新的联合治疗提供重要线索。
纪念斯隆-凯特琳癌症中心的梁德光博士是本文的第一作者,姜学军教授是该研究的通讯作者。此外,该项研究工作得到了哥伦比亚大学Brent R. Stockwell教授和Wei Gu教授等合作者的大力支持。

