一个能加速癌症免疫治疗发展的研究成果出现了!
近日,由乔治·华盛顿大学的Li Rong和德克萨斯大学的An Zhiqiang等领衔的研究团队,在顶级期刊《自然》发表重磅研究成果[1]。他们发现,在癌症进展过程中,一种被称为DDR1的分子会释放其胞外结构,组织胶原纤维在肿瘤周边形成致密的免疫屏蔽结构,阻止免疫细胞进入肿瘤。而特异性抑制DDR1胞外结构的活性,免疫细胞可以渗入肿瘤,并杀死里面的癌细胞。An Zhiqiang等认为,发现DDR1在癌症耐药性中的重要作用是一项重大进展,有可能改变癌症治疗途径[2]。

在三阴性乳腺癌(TNBC)等多种恶性肿瘤中,免疫排斥与患者的不良预后有关[3]。已经有很多研究表明,细胞外基质(ECM)有助于免疫排斥的发生[4]。然而,减少ECM的策略要么无效,要么会产生不希望看到的结果[5,6]。Li Rong团队注意到,有一个叫做盘状结构域受体1(DDR1)的受体酪氨酸激酶,它的表达增加与包括乳腺癌在内的多种癌症的进展相关[7]。遗憾的是,DDR1促进肿瘤进展和转移的机制,还知之甚少[8,9]。不过,我们知道的是,DDR1在细胞的黏附,增殖及细胞外基质的重塑中起重要作用。因此Li Rong团队将DDR1作为研究的重点。为了研究DDR1的促癌机制,Li Rong团队在三种TNBC小鼠模型(E0771、M-Wnt和AT-3)中,特异性删除了Ddr1基因。他们发现,Ddr1的敲除(KO)不损害癌细胞在体外的增殖,也不影响癌细胞在免疫缺陷宿主体内生长。不过,Ddr1-KO肿瘤在免疫功能正常的宿主体内不能生长。如果耗尽CD8+细胞的话,Ddr1-KO肿瘤就能正常生长,长势与对照的野生型(Ddr1-WT)肿瘤一样旺盛。
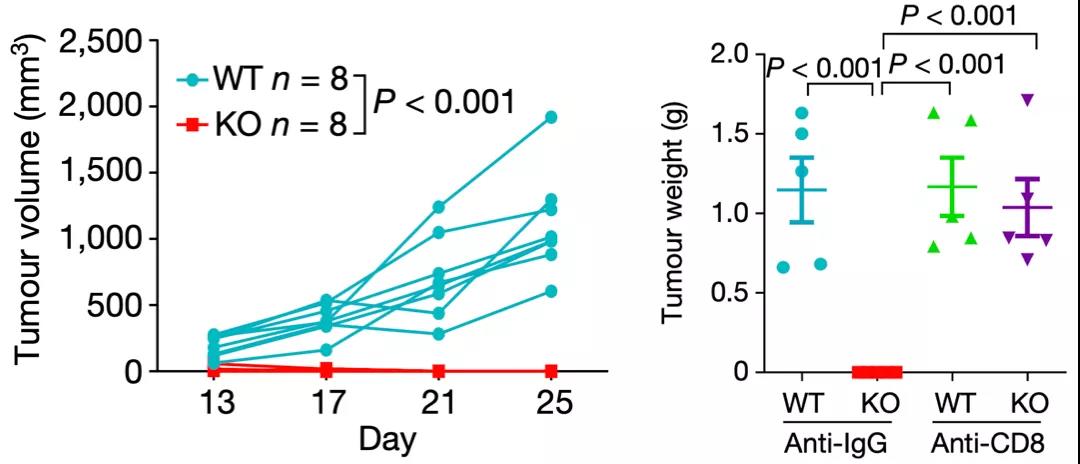
Ddr1-KO肿瘤在免疫功能正常的宿主体内不能生长,耗尽CD8+细胞后正常生长
还有个让研究人员意外的发现是:将CD8+ T细胞分别转移到免疫缺陷的荷瘤小鼠体内,与野生型Ddr1-WT肿瘤相比,更多的CD8+ T细胞浸润到了Ddr1-KO肿瘤中。而且,原本在免疫缺陷鼠身上一样茁壮生长的Ddr1-KO和Ddr1-WT肿瘤,变得大小不一样了,Ddr1-KO明显变小了。这些结果表明,DDR1阻碍了小鼠的抗肿瘤免疫能力。进一步的分析之后,Li Rong团队发现,原来DDR1仅仅是阻碍了T细胞的浸润,而不是影响T细胞的增殖或细胞毒性功能。更直观地看,Ddr1-WT肿瘤将CD8+ T细胞拒之门外,更多的CD8+ T细胞聚集在肿瘤的边缘;而Ddr1-KO肿瘤的核心被更多的CD8+ T细胞渗透、浸润。这进一步证明:DDR1限制了T细胞的浸润。
为了验证以上的发现,Li Rong团队又分析了一些临床样本。他们发现,与肿瘤边缘相比,DDR1水平高的肿瘤内部的CD8+ T细胞丰度较低。相比之下,DDR1低的肿瘤在肿瘤核心和肿瘤边缘的CD8+ T细胞丰度方面没有任何明显的差异。
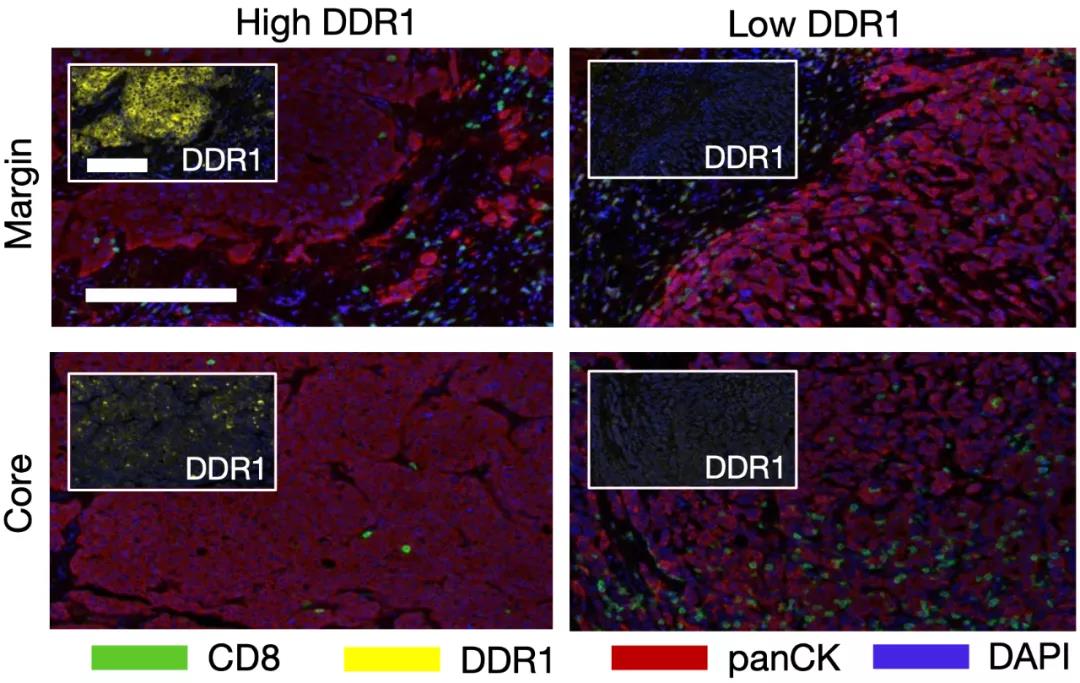
DDR1限制了T细胞的浸润
而且,当这个队列按核心和边缘的相对CD8+ T细胞密度分层时,所有具有免疫排斥表型的肿瘤都是DDR1水平高,而大多数非免疫排斥的肿瘤是DDR1水平低。这些临床数据共同支持了他们的临床前发现:DDR1排除了抗肿瘤免疫细胞。那么CD8+ T细胞为啥不能进到DDR1高水平的肿瘤内部呢?Li Rong团队打算从头捋起。一般来讲,CD8+ T细胞浸润肿瘤有三关:血管外渗、肿瘤诱导的趋化作用和穿越细胞外基质(ECM)。将Ddr1-WT肿瘤和Ddr1-KO肿瘤对比一番之后,Li Rong和他的同事发现:二者的血管无甚差异;肿瘤本身除了DDR1水平之外,在趋化因子和抗原等方面也没有差异。为了进一步挖掘信息,研究人员甚至还比较了Ddr1-WT肿瘤和Ddr1-KO肿瘤,分别在免疫缺陷小鼠、免疫正常小鼠和体外条件下的转录组差异。汇总所有的信息之后,Li Rong和他的同事发现,所有的信息似乎都指向了肿瘤基质细胞。或许该研究研究DDR1缺失对这个群体的影响了。遗憾的是,他们没有发现DDR1缺失对肿瘤基质细胞有显著的影响。看来还是得回到DDR1本身。

Li Rong教授(图源:乔治·华盛顿大学)
前面我们介绍过,DDR1是一个跨膜的受体酪氨酸激酶,它的胞外结构域(DDR1-ECD)能与胶原蛋白结合,触发下游的信号传导事件[10]。既然毫无头绪,那就一点点研究DDR1的各部分功能。Li Rong团队注意到,在Ddr1-KO肿瘤中表达全长的DDR1或删除了激酶结构域的DDR1(ΔKD),都能恢复Ddr1-KO肿瘤在免疫正常小鼠体内的生长。值得注意的是,同时缺乏跨膜和激酶结构域的DDR1-ECD,竟然也能支持Ddr1-KO肿瘤的生长。不难看出,DDR1结合胶原蛋白的能力,极有可能是促进肿瘤生长的关键。为了确认这一发现,Li Rong和他的同事给DDR1-ECD引入基因突变。他们发现,那些没有损害DDR1-ECD结合胶原蛋白能力的变异,不会损害DDR1-ECD促进肿瘤生长的能力;而破坏了DDR1-ECD结合胶原蛋白能力的变异,会导致DDR1-ECD促进肿瘤生长的能力大幅下降。
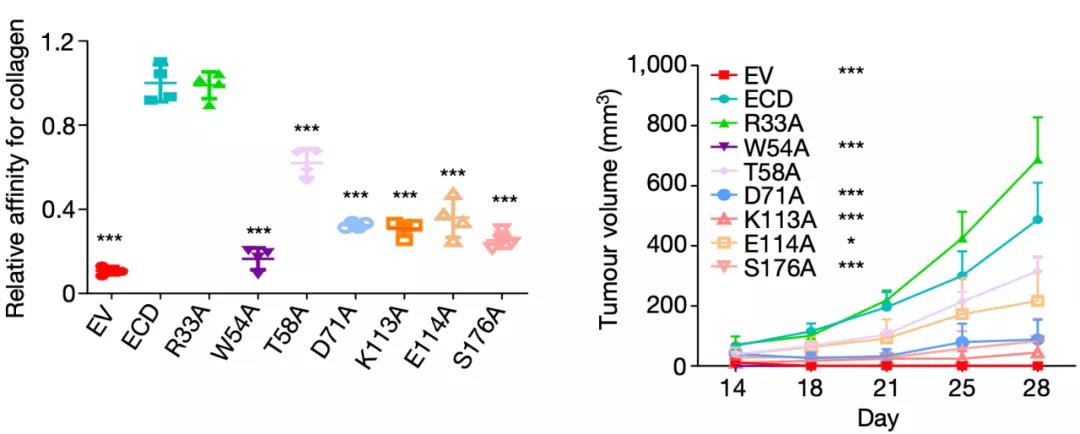
不同的变异对肿瘤生长的影响
总的来说,以上数据表明,DDR1的胶原蛋白结合能力——而不是其激酶活性——是促进肿瘤在免疫能力强的小鼠体内生长所必需的。那么DDR1的胞外结构域(DDR1-ECD)又是如何帮助肿瘤屏蔽免疫细胞的呢?这个时候,Li Rong和他的同事注意到,之前已经有研究发现表达DDR1的细胞会脱落DDR1-ECD[11],而且多聚体形式的DDR1-ECD能重塑胶原纤维[12]。但是这一现象的生物学意义仍不为人知。或许这就是突破口。通过进一步观察,Li Rong团队发现,Ddr1-WT肿瘤边缘的胶原纤维长而整齐,而Ddr1-KO肿瘤的胶原纤维短、不整齐,且分散。神奇的是,在免疫缺陷小鼠身上,Ddr1-WT肿瘤和Ddr1-KO肿瘤的胶原纤维,无论是长度还是排列都没有差异。这意味着胶原纤维的这种变化,是肿瘤与免疫系统之间交战的结果。
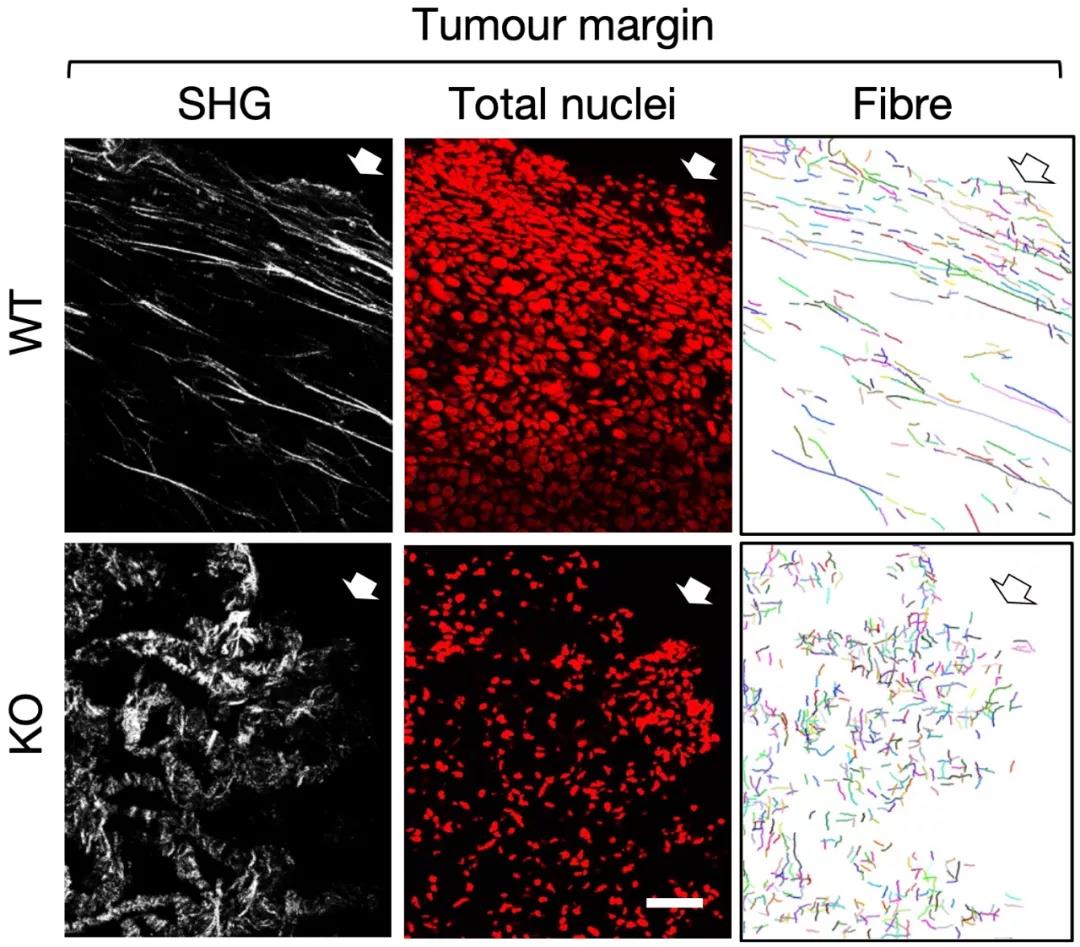
注意看纤维的差异
随后,Li Rong和他的同事发现,瘤内注射正常的DDR1胞外结构域能增强胶原蛋白纤维的排列,抑制免疫细胞浸润;而注射DDR1胞外结构域的功能缺失突变体就没有这个效果。现在,所有问题都搞清楚啦。就拿本研究关注的三阴性乳腺癌来说,肿瘤与免疫系统的交锋,可能促进了DDR1胞外结构域的脱落,胞外结构域一手操办了胶原纤维的排列,给肿瘤打造了一个“刀枪不入”的免疫屏蔽场。
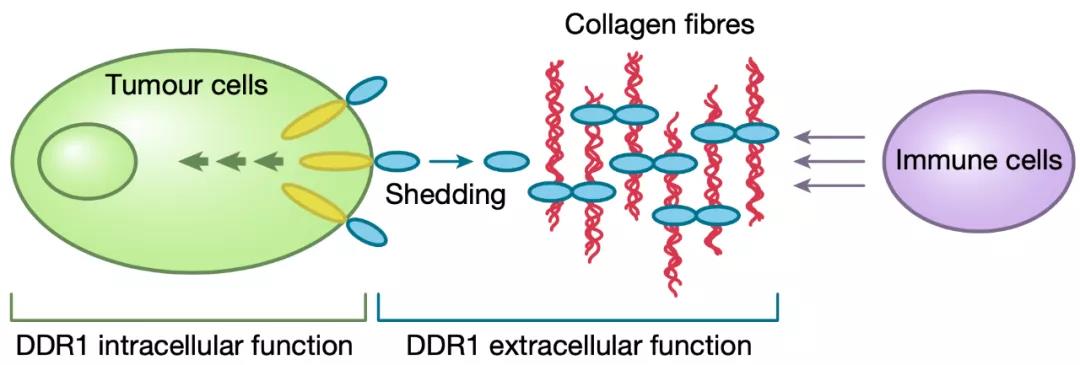
机制插图
在研究的最后,Li Rong团队还筛选了靶向DDR1胞外结构域的特异性单克隆抗体,证实在肿瘤形成之后,特异性抑制DDR1胞外结构域的活性,有良好的抗肿瘤活性,甚至能让肿瘤完全消失。那些被特异性单克隆抗体处理过的肿瘤免疫细胞浸润增多,肿瘤边缘的胶原纤维排列不整齐、较短。这再次证实了前面的发现。不难看出,Li Rong团队的这个研究可谓意义重大。因为很多肿瘤都是没有免疫浸润的冷肿瘤,免疫治疗都拿它们没有办法。如果能用DDR1胞外结构域的特异性抗体打破肿瘤的免疫“屏蔽场”,再辅以免疫检查点抑制剂,或许很多冷肿瘤的治疗难题将迎刃而解。据了解,研究团队已经将这一研究成果授权给Parthenon Therapeutics。在研究成果发表的同一天,Parthenon Therapeutics宣布获得了6500万美元A轮融资,用于相关药物PRTH-101的研发[13]。
无论如何,期待这个研究早日转化到临床,造福更多的癌症患者。
参考文献:
[1].Sun, X., Wu, B., Chiang, HC. et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature (2021). https://doi.org/10.1038/s41586-021-04057-2
[2].https://gwtoday.gwu.edu/gw-researchers-identify-molecule-blocks-immune-cells-killing-breast-tumors
[3].Gruosso T, Gigoux M, Manem VSK, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest. 2019;129(4):1785-1800. doi:10.1172/JCI96313
[4].Cox TR. The matrix in cancer. Nat Rev Cancer. 2021;21(4):217-238. doi:10.1038/s41568-020-00329-7
[5].Bejarano L, Jordāo MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021;11(4):933-959. doi:10.1158/2159-8290.CD-20-1808
[6].Chen Y, Kim J, Yang S, et al. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39(4):548-565.e6. doi:10.1016/j.ccell.2021.02.007
[7].Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31(1-2):295-321. doi:10.1007/s10555-012-9346-z
[8].Hidalgo-Carcedo C, Hooper S, Chaudhry SI, et al. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol. 2011;13(1):49-58. doi:10.1038/ncb2133
[9].Gao H, Chakraborty G, Zhang Z, et al. Multi-organ Site Metastatic Reactivation Mediated by Non-canonical Discoidin Domain Receptor 1 Signaling. Cell. 2016;166(1):47-62. doi:10.1016/j.cell.2016.06.009
[10].Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. 2014;310:39-87. doi:10.1016/B978-0-12-800180-6.00002-5
[11].Vogel WF. Ligand-induced shedding of discoidin domain receptor 1. FEBS Lett. 2002;514(2-3):175-180. doi:10.1016/s0014-5793(02)02360-8
[12].Agarwal G, Mihai C, Iscru DF. Interaction of discoidin domain receptor 1 with collagen type 1. J Mol Biol. 2007;367(2):443-455. doi:10.1016/j.jmb.2006.12.073
[13].https://www.businesswire.com/news/home/20211103005945/en/Parthenon-Therapeutics-Raises-65-Million-in-Series-A-Funding-to-Advance-Oncology-Programs-Aimed-at-Reprogramming-the-Tumor-Microenvironment

